Chemprop: A Machine Learning Package for Chemical Property Prediction
Published in ChemRxiv, 2023
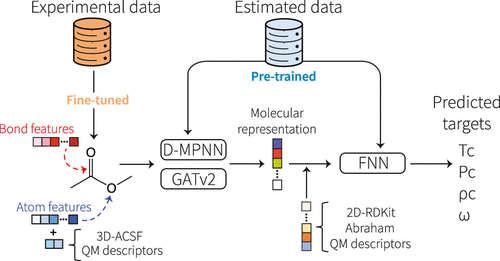
Deep learning, particularly directed message-passing neural networks (D-MPNNs), has emerged as a robust method for predicting molecular properties. Chemprop, an open-source software, employs the D-MPNN architecture to provide swift and straightforward access to machine-learned molecular properties. In its latest iteration, Chemprop boasts added functionalities like support for multi-molecule properties, reactions, atom/bond-level properties, and spectra. The package also incorporates uncertainty quantification, transfer learning, enhanced hyperparameter optimization, and other tailored options. Benchmarked on datasets like MoleculeNet and SAMPL, Chemprop has demonstrated state-of-the-art performance in predicting various molecular attributes. The software simplifies the process of training D-MPNN models, ensuring efficiency and user-friendliness.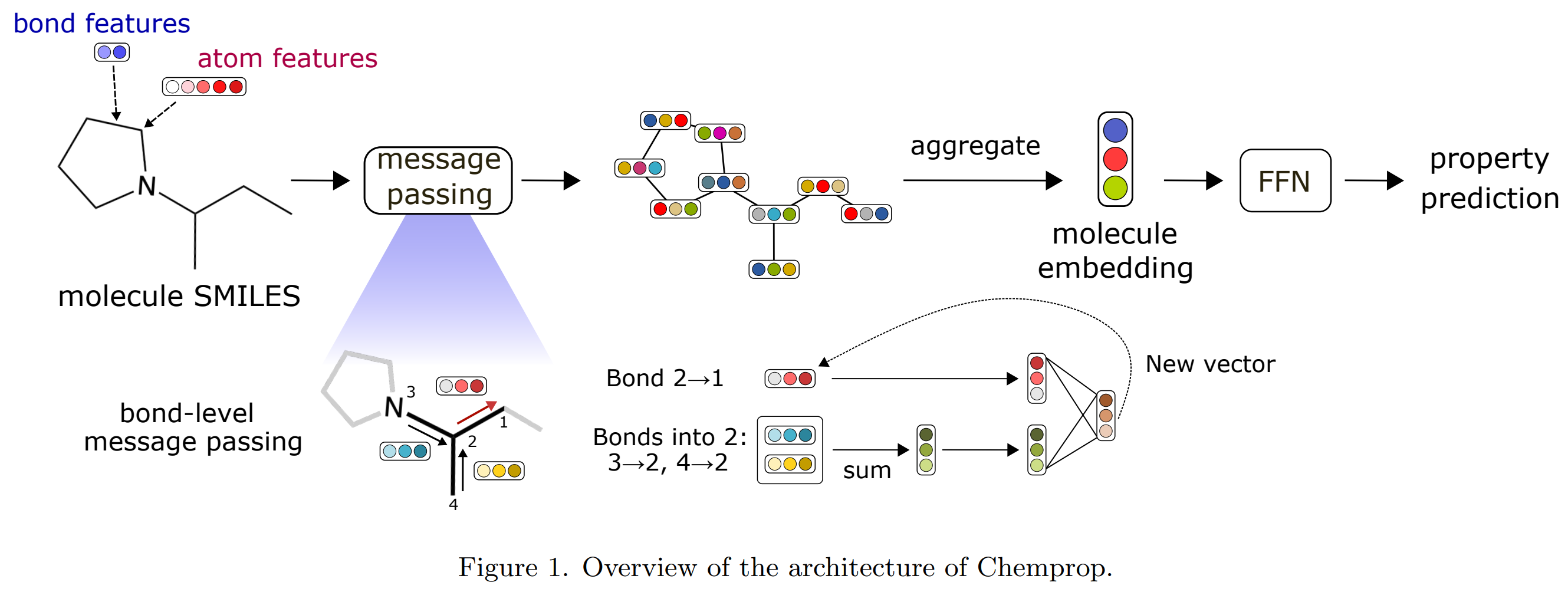
Recommended citation: Heid, Esther; Greenman, Kevin P; Chung, Yunsie; Li, Shih-Cheng; Graff, David E; Vermeire, Florence H; Wu, Haoyang; Green, William H; McGill, Charles J. (2023). "Chemprop: A Machine Learning Package for Chemical Property Prediction." https://chemrxiv.org/engage/chemrxiv/article-details/64d1f13d4a3f7d0c0dcd836b